#17,463
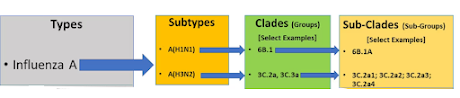
With 18 HA (hemagglutinin) subtypes and 11 different NA (neuraminidase) subtypes there are nearly 200 possible influenza subtypes (e.g. H1N1, H5N1, H7N8, etc.). To date, more than 130 have already been detected in nature.
Each subtype (based on the HA gene) is further classified by its clade, and within each clade there can be multiple subclades (see chart below). This alone can yield thousands of variations.

In order to thrive, a new subtype/clade/genotype must be able to compete against the existing panoply of flu viruses, and luckily most are evolutionary failures. But occasionally a more biologically `fit' virus emerges, often with dramatic effect.
We saw that happen in 2005 at Qinghai Lake, China where a new clade (2.2) emerged, followed by the sudden spread of H5N1 into Europe, the Middle East, and North Africa. Other emerging clades (2.3.3 and 2.3.2.1c) helped propel H5N1 into India, Japan, and West Africa.
By the middle of the last decade HPAI H5 appeared to be on the decline, at least until a reassortment event occurred somewhere around Ubsu-Nur Lake in Russia over the summer of 2016. Prior to that, HPAI H5 viruses were poorly adapted for carriage by many bird species.
Changes picked up during this event allowed the virus to persist in more bird species, sparking the biggest avian epizootic (to that date) in Europe over the 2016/2017 winter season.
Since then, the virus has undergone many changes, including different subtypes (H5N6, H5N5, H5N1), and has used its ability to spread more readily via migratory birds to expand its range to the Americas.
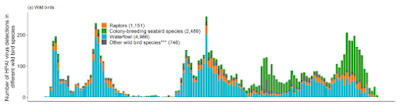
Evolving `Order Shift' of Birds Detected With HPAI H5
Like an avalanche rolling down a mountainside, the farther the virus travels and the more diverse it becomes, the more momentum it gains. New genotypes are being reported on a regular basis, and there are undoubtedly many in the wild that we aren't even aware of.
And of course, it isn't just viral evolution in birds. Mammals are increasingly being infected (see below), adding new evolutionary pathways to the mix.
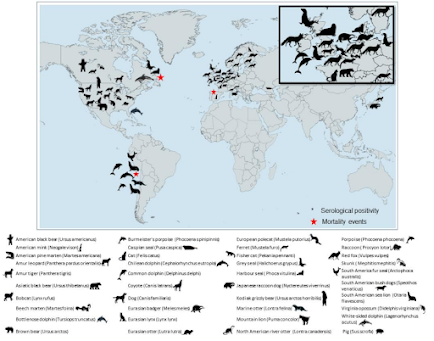
We are essentially witnessing a massive unregulated global GOF (Gain of Function) experiment, one that continues to expand with every passing day. While individually, these H5 variants are all a long-shot to generate a pandemic virus, the huge number of rolls of the genetic dice each day does not work in our favor.
Which is why reports, such as the EID Journal Research Letter below, are important.
The discovery of additional genotypes of H5N1 in South Korea may not be as newsworthy as suspected human infections in Brazil, or the die off of seals in New England, but each new iteration of the HPAI H5 virus is a potential stepping stone towards the generation of the next pandemic virus.
The authors of today's Research letter report finding several mutations in five H5N1 samples linked to mammalian adaptation of the virus. They also provide a brief overview of the evolution of the H5 virus over the past decade.
I've only posted some excerpts, so follow the link to read the report in its entirety.
Research Letter
Novel Highly Pathogenic Avian Influenza A(H5N1) Clade 2.3.4.4b Virus in Wild Birds, South Korea
Sun-hak Lee, Andrew Y. Cho, Tae-hyeon Kim, Seo-jeong Ahn, Ju Ho Song, Heesu Lee, Yun-Jeong Choi, Nyamsuren Otgontogtokh, Jung-Hoon Kwon, Chang-Seon Song, and Dong-Hun Lee
Abstract
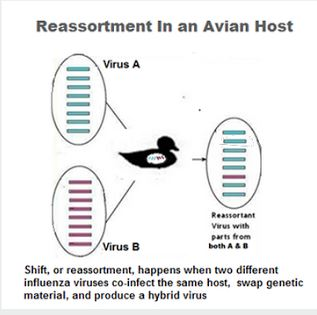
We isolated 5 highly pathogenic avian influenza A(H5N1) clade 2.3.4.4.b viruses from wild waterfowl feces in South Korea during November 2022. Whole-genome sequencing and phylogenetic analysis revealed novel genotypes produced by reassortment with Eurasian low pathogenicity avian influenza viruses. Enhanced surveillance will be required to improve prevention and control strategies.
Highly pathogenic avian influenza viruses (HPAIVs) have caused major economic losses in the poultry industry and are a major threat to public health. Since the first detection of HPAIV A(H5N1) from a goose in 1996 in Guangdong, China, its descendants have evolved into multiple hemagglutinin (HA) gene-specific clades (H0–H9) and subclades (1) causing intercontinental epizootics (2). Over several decades, H5 HPAIVs have evolved into multiple subtypes and genotypes generated by reassortment with low pathogenicity avian influenza viruses (LPAIVs), which led to emergence of clade 2.3.4.4 H5Nx HPAIVs in eastern China during 2013–2014 (3).
In mid-2016, reassortant H5N8 clade 2.3.4.4b HPAIVs that contained internal genes of LPAIVs from Eurasia were detected in wild birds at Uvs-Nuur Lake in Russia and Qinghai Lake in China (4); the viruses caused large outbreaks in Europe during 2016–2017 (5). Subsequently, various novel reassortant H5N8 HPAIVs were detected in Eurasia (5,6). In late 2020, novel reassortant clade 2.3.4.4b H5N1 HPAIVs were detected and became predominant in Europe in poultry and wild birds (5).
(SNIP)The H5N1 HPAIVs from South Korea contained amino acids in HA with binding affinity for avian α-2,3-linked sialic acid receptors (T118, V210, Q222, and G224) (H5 numbering) (8,9). They also had 2 HA amino acid substitutions, S113A and T156A, associated with increased binding affinity to human α-2,6-linked sialic acid receptors (Appendix 1 Table 2).
All 5 isolates had amino acid substitutions that included A515T in PA, known to increase polymerase activity in mammal cells, and N30D, I43M, T215A in MP1 and L89V in PB2, known to increase virulence in mice (Appendix 1 Tables 2, 3).
The HPAI/LPAI reassortment of H5Nx clade 2.3.4.4b HPAIVs created a diverse genetic pool of H5 clade 2.3.4.4 viruses that continuously emerged in various countries (1). Clade 2.3.4.4 H5N8 HPAIV isolated from Uvs-Nuur Lake in Russia had reassorted H3N8 LPAIV genes from Mongolia (4).
In Europe, HPAIVs identified in 2020 (5,6) were produced by reassortment between clade 2.3.4.4b HPAIV and LPAIVs from Eurasia. Novel reassortments of clade 2.3.4.4 HPAIV and LPAIVs from Eurasia were also detected in 2016 (10), during 2020–2021 (Appendix 1 reference 1), and in late 2021 (Appendix 1 reference 2) in South Korea.
Considering the continuous emergence and global dissemination of novel reassortant clade 2.3.4.4b HPAI H5Nx viruses, enhanced active surveillance in wild animals and domestic poultry will be required to monitor the introduction, dissemination, and evolution of HPAIVs and provide insight for improved prevention and control strategies.
Mr. Lee is a PhD candidate at Konkuk University, Seoul, South Korea. His primary research interests focus on molecular epidemiology and host–pathogen interactions of avian influenza viruses.
While we may find there is some as-yet unidentified `species barrier' that would prevent H5N1 from sparking a pandemic (see Are Influenza Pandemic Viruses Members Of An Exclusive Club?), there is little to prevent H5N1 from reassorting with seasonal H1N1 or H3N2, or with any of dozens of other H1, H2, or H3 viruses in the wild.
Viruses have been finding a way to survive for millions of years.
We underestimate them at our own peril.