#17,111
It is not just a theoretical concern. We've already seen it happen.
In the fall of 2020 SARS-CoV-2 jumped from humans to farmed mink in Denmark, and began to mutate into new mink-variants (see Denmark Orders Culling Of All Mink Following Discovery Of Mutated Coronavirus). As a result, several mutated viruses jumped back into humans (see WHO 2nd Update: SARS-CoV-2 mink-associated variant strain – Denmark).
Luckily, this emergency was short-lived, as the Alpha variant emerged in Europe in late 2020 and quickly supplanted these mink-variants. But it did demonstrate the problem; carriage of SARS-CoV-2 by other host species can produce new variants, which can jump back into humans.
Since then, we've seen similar events in the United States (see CDC: Investigating Possible Mink-To-Human Transmission Of SARS-CoV-2 In The United States) and in Hong Kong (see Hong Kong Detects COVID In Pet Store Hamsters - Suspends Sales & Orders Cull).
Regardless of Omicron's path to global domination, the threat is real. Which is why seven months ago we saw the WHO/FAO/OIE Joint Statement On Monitoring SARS-CoV-2 In Wildlife & Preventing Formation of Reservoirs.
A few (of many) past blogs on SARS-CoV-2 spillover to non-human species includes:
Preprint: SARS-CoV-2 Infection in Domestic Rats After Transmission From Their Infected Owner
USDA/APHIS: White-Tailed Deer Exposed To SARS-CoV-2 Detected In 4 States
EID Journal: Peridomestic Mammal Susceptibility to SARS-CoV-2 Infection
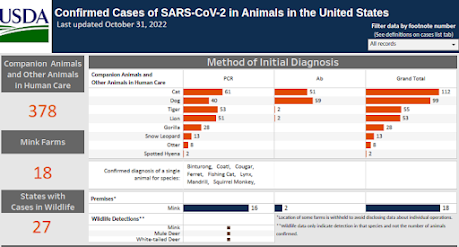
The USDA maintains a dashboard showing the number of confirmed non-human infections with SARS-CoV-2 in the United States (see below), but this undoubtedly only represents a tiny fraction of cases, and only covers the United States.
Wildlife exposure to SARS-CoV-2 across a human use gradientAmanda R Goldberg,Kate Elizabeth Langwig, Jeffrey Matthew Marano, Amanda K Sharp, Katherine L Brown, Alessandro Ceci, Macy J Kailing, Russell Briggs, Clinton Roby, Anne M Brown, VJames Weger-Lucarelli, Carla v Finkielstein, Joseph R Hoyt
doi: https://doi.org/10.1101/2022.11.04.515237
Abstract
The spillover of SARS-CoV-2 into humans has caused one of the most devastating pandemics in recorded history. Human-animal interactions have led to transmission events of SARS-CoV-2 from humans to wild and captive animals. However, many questions remain about how extensive SARS-CoV-2 exposure is in wildlife, the factors that influence wildlife transmission risk, and whether sylvatic cycles can generate novel variants with increased infectivity and virulence.We sampled 18 different wildlife species in the Eastern U.S. and detected widespread exposure to SARS-CoV-2 across wildlife species. Using quantitative reverse transcription polymerase chain reaction and whole genome sequencing, we conclusively detected SARS-CoV-2 in the Virginia opossum and had equivocal detections in six additional species.Species considered human commensals like squirrels, and raccoons had high seroprevalence, ranging between 62%-71%, and sites with high human use had three times higher seroprevalence than low human-use areas.SARS-CoV-2 genomic data from an infected opossum and molecular modeling exposed previously uncharacterized changes to amino acid residues observed in the receptor binding domain (RBD), which predicts improved binding between the spike protein and human angiotensin-converting enzyme (ACE2) compared to the dominant variant circulating at the time of isolation. These mutations were not identified in human samples at the time of collection.Overall, our results highlight widespread exposure to SARS-CoV-2 in wildlife and suggest that areas with high human activity may serve as important points of contact for cross-species transmission. Furthermore, this work highlights the potential role of wildlife in fueling de novo mutations that may eventually appear in humans.
(SNIP)
Discussion
We found that exposure to SARS-CoV-2 appears widespread among wildlife species. We detected positives in 6 species using RT-qPCR and neutralizing antibodies in an additional 4 species. We found support for a relationship between human use and seroprevalence suggesting that areas with high human activity are potential hotspots for cross-species transmission.
Analysis of isolates collected from infected wildlife revealed novel mutations at the time of collection which have subsequently been found circulating in other regions. While some of these mutations likely increase binding affinity to the hACE2 receptor or confer some antibody resistance compared to ancestral BA.2 lineages, others appeared unique to the opossum. Whether these mutations developed in humans not captured in surveillance at the time or in wildlife communities and were transmitted back to humans remains unknown but presents an important consideration for future variant surveillance.
(SNIP)
SARS-CoV-2 isolated from the opossum, showed unique mutations in the spike protein in the RBM and RBD of the spike protein. These previously uncharacterized amino acid changes are predicted to improve S-hACE2 binding and membrane fusion efficiencies compared to omicron BA.2, potentially providing a fitness advantage by either increasing the affinity of S for the ACE2 receptor or, alternatively, by evading the neutralizing activity of antibodies. These mutations were not observed in human samples at the time of isolation from the opossum and have only recently been identified in a new Omicron sub-variant (BJ.1). These results highlight the role of wildlife in contributing unique mutations that may be more transmissible to humans as has been previously suggested with the emergence of the Omicron variant (6, 24)
How often SARS-CoV-2 transmits to animals in the wild is largely unknown, since only a few narrowly targeted surveillance programs have been attempted. And while most de novo mutations will end up being evolutionary failures, it only takes one biological over-achiever to reignite the pandemic.
Which means the next global public health crisis may be much closer than we think.A little over a year ago PNAS Research: Intensity and Frequency of Extreme Novel Epidemics, researchers suggested that the probability of novel disease outbreaks will likely grow three-fold in the next few decades.