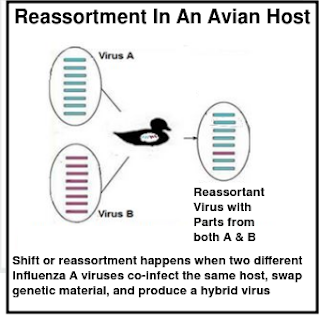
This evolutionary process is called reassortment (or antigenic shift).
Reassortment can lead to the abrupt creation of new hybrid viruses (either new subtypes or new genotypes) and is the most likely way that a pandemic flu virus will emerge (see 3 min. NIAID Video: How Influenza Pandemics Occur).
Viruses can also evolve through a much slower process called antigenic drift (aka replication errors), which can accelerate when a flu virus spills over into a new species (aka `host adaptation').
The bottom line is the more H5N1 spreads, the more opportunities it will have to reinvent itself. Each new spillover is another opportunity to adapt (via `drift' and/or host adaptation) and each coinfection with another influenza A virus is another opportunity to reassort into a new genotype (or subtype).
While most of these reassortants will be evolutionary failures - unable to compete with their more biologically `fit' parental strains - with the virus granted millions of opportunities on its world tour, an occasional success story is inevitable.
We've already seen several new genotypes reported over the past year as H5N1 clade 2.3.4.4b viruses have swept across Europe and North America (see Preprint: Rapid Evolution of A(H5N1) Influenza Viruses After Intercontinental Spread to North America) and as one might expect, we are seeing new genotypes emerge in South America now as well.
All of which brings us to a new preprint, published yesterday on the bioRxiv, which describes the evolution new genotypes of avian H5N1 clade 2.3.4.4b as it spreads through wild birds and marine mammals in Peru.
This is lengthy (31-page PDF) report, with a lot of technical details. I've only posted the abstract below, along with some brief excerpts from the discussion. Follow the link to read it in its entirety.
Highly pathogenic avian influenza A (H5N1) in marine mammals and seabirds in Peru
Mariana Leguia, Alejandra Garcia-Glaessner, Breno Munoz-Saavedra, Diana Juarez, Patricia Barrera, Carlos Calvo-Mac, Javier Jara, Walter Silva, Karl Ploog, Lady Amaro, Paulo Colchao-Claux, Marcela M Uhart, Martha I Nelson, Jesus Lescano
doi: https://doi.org/10.1101/2023.03.03.531008
Abstract
Highly pathogenic avian influenza (HPAI) A/H5N1 viruses (lineage 2.3.4.4b) are rapidly invading the Americas, threatening wildlife, poultry, and potentially evolving into the next global pandemic.In November 2022, HPAI arrived in Peru, where massive pelican and sea lion die-offs are still underway. We report complete genomic characterization of HPAI/H5N1 viruses in five species of marine mammals and seabirds (dolphins, sea lions, sanderlings, pelicans and cormorants) sampled since November 2022.All Peruvian viruses belong to the HPAI A/H5N1 lineage 2.3.4.4b, but they are 4:4 reassortants where 4 genomic segments (PA, HA, NA and MP) position within the Eurasian lineage that initially entered North America from Eurasia, while the other 4 genomic segments (PB2, PB1, NP and NS) position within the American lineage (clade C) that was already circulating in North America.These viruses are rapidly accruing mutations as they spread south. Peruvian viruses do not contain PB2 E627K or D701N mutations linked to mammalian host adaptation and enhanced transmission, but at least 8 novel polymorphic sites warrant further examination.This is the first report of HPAI A/H5N1 in marine birds and mammals from South America, highlighting an urgent need for active local surveillance to manage outbreaks and limit spillover into humans.
(SNIP)
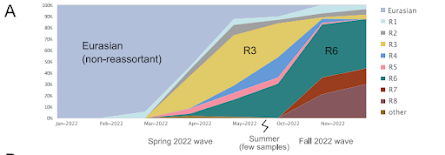
Figure 3: Spread of reassortant “R6” viruses. (A) Proportion of H5N1 viruses sequenced in the Americas between 2021-2023 that belong to different genotypes (Table 2) over time
There are outstanding questions about which migratory bird species are involved in the long distance dissemination of HPAI from North to South America, possibly by way of Central America. We detected clade 2.3.4.4b in a migratory sanderling (Calidris alba) that would have arrived in Peru after breeding in the Canadian arctic. However, Calidris spp. are an unlikely conduit for HPAIV A/H5N1 because experimental inoculations result in death or disease within 5 to 11 days of inoculation37,38 .
Given the unlikelihood of a successful long-distance migration for a clinically infected bird, we suspect the sanderling was infected locally. Our phylogenetic analysis supports multiple independent introductions of HPAI from North America into South American countries for which sequence data was available at the time of this study, including Peru, Ecuador, and Venezuela. This contrasts with the single introduction of HPAI from Eurasia to North America observed earlier in 2021.
Although North America is the primary source of HPAI for South America’s initial HPAI outbreaks, South American countries are likely to become more important sources for each other’s HPAI outbreaks as the virus establishes locally. After observing HPAI A/H5N1 reassort repeatedly within North American viruses, it is possible that the virus will continue to evolve in South America by mutation and reassortment with the genetically distinct South American AIV lineage that is commonly detected in Argentina39 and Chile40 .
In addition to the 40 previously characterized variable sites linked to concerning phenotypes that we observed in the 8 Peruvian HPAI A/H5N1 genomes, we identified an additional 30 sites that remain uncharacterized. There is an urgent need to establish pipelines for efficient real-time genomic sequencing of HPAI to track its evolution as it spreads across Peru and other countries in South America, as well as funding to support characterization of possible new mutations.
While the generation of a pandemic virus from H5N1 clade 2.3.4.4b is undoubtedly a statistical long shot, similar evolutionary processes are going on for all influenza A viruses (avian, swine, human, canine, etc.) around the globe, most of which are hidden from our view.
Even if clade 2.3.4.4b H5N1 is ultimately shown not to have the `right stuff' to spark a human pandemic, somewhere out there is a influenza A virus - promiscuously spreading, and reassorting, in a pig, a bird, a seal, a cat, or even a bat - which does.
History has shown that these pandemics have emerged at irregular intervals for the past 500 years, and there is no reason to believe they will stop anytime soon.