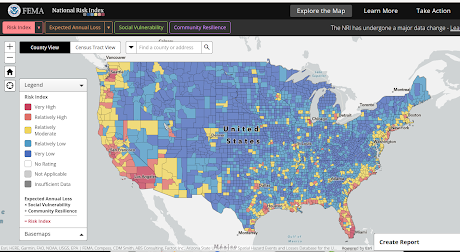
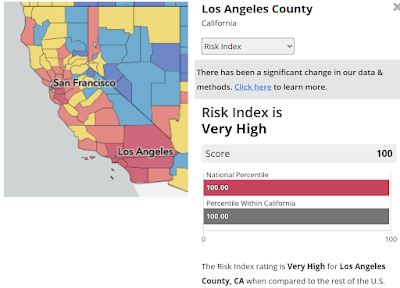
Credit ACIP/CDC
#17,380
Although COVID vaccines have undoubtedly saved millions of lives, and spared countless more from severe illness, one of the theoretical concerns over the deployment of any pandemic vaccine is that they might help drive viral evolution, leading to the generation of vaccine-escape variants.
There is some evidence to suggest that the use of (often substandard, or poorly matched) poultry vaccines has led to the rise of new new, potentially more dangerous, avian flu strains.
The following comes from a 2014 EID Journal dispatch titled Subclinical Highly Pathogenic Avian Influenza Virus Infection among Vaccinated Chickens, China:
HPAI mass vaccination played a crucial role in HPAI control in China. However, this study demonstrated multiple disadvantages of HPAI mass vaccination, which had been suspected (13,14). For example, this study showed that H5N1 subtype HPAI virus has evolved into multiple H5N2 genotypes, which are all likely vaccine-escape variants, suggesting that this virus can easily evolve into vaccine-escape variants.
This observation suggests that HPAI mass vaccination, which is highly effective in the beginning of an outbreak, may lose its effectiveness with time unless the vaccine strains are updated. Moreover, this study showed that vaccinated chicken flocks can be infected with vaccine-escape variants without signs of illness.
We know that vaccines don’t always prevent infection. Sometimes they only mask or minimize the symptoms, and in that environment vaccine resistant mutations may emerge and potentially be transmitted onward.
This is a concern we explored in the summer of 2021 (see UK Sage: International Vaccination: Potential impact on Viral Evolution and UK), in a report that looked at the potential impact of vaccine-induced viral evolution of SARS-CoV-2.
While the authors argued that increased international vaccine coverage was more likely to help prevent the emergence of new variants, they acknowledged there were unknowns.
At roughly the same time, in UK SAGE: Can We Predict the Limits of SARS-CoV-2 Variants and their Phenotypic Consequences?), that expert group called additional genetic and antigenic drift of the virus almost inevitable. As predicted, since then we've seen a remarkable expansion in the diversity of SARS-CoV-2 viruses, including the emergence of the (now dominant) Omicron lineage.
But the question remains: Did the COVID vaccine contribute to this abrupt evolutionary shift in the virus?
Last fall, in Viruses: Evolutionary Pattern Comparisons of the SARS-CoV-2 Delta Variant in Countries/Regions with High and Low Vaccine Coverage, we looked at a study - based on limited, country-level data - that found little evidence that high vaccine uptake was driving the evolution of the SARS-CoV-2 virus.
Today we've another look at the issue from some well-known names in virology from the University of Hong Kong.
Instead of comparing the emergence of variants from high and low vaccine uptake countries, they actually looked at the diversity of COVID strains detected within thousands of vaccinated and unvaccinated individuals.
This is, as you might imagine, a lengthy, detailed, and highly technical report. I've only reproduced the Abstract, and some excerpts from the Introduction and Discussion.
Those without a high tolerance for statistics and a basic understanding of virology will find it tough sledding (I know I did), but the whole report is very much worth reading.
While this may not be the final word on the subject, reassuringly these researchers did not find any evidence suggesting vaccinated individuals are more likely to produce, and spread, viral variants.
Article
Open Access
Published: 31 March 2023
Within-host genetic diversity of SARS-CoV-2 lineages in unvaccinated and vaccinated individuals
Haogao Gu, Ahmed Abdul Quadeer, Pavithra Krishnan, Daisy Y. M. Ng, Lydia D. J. Chang, Gigi Y. Z. Liu, Samuel M. S. Cheng, Tommy T. Y. Lam, Malik Peiris, Matthew R. McKay & Leo L. M. Poon
Nature Communications volume 14, Article number: 1793 (2023) Cite this article
Abstract
Viral and host factors can shape SARS-CoV-2 evolution. However, little is known about lineage-specific and vaccination-specific mutations that occur within individuals. Here, we analysed deep sequencing data from 2,820 SARS-CoV-2 respiratory samples with different viral lineages to describe the patterns of within-host diversity under different conditions, including vaccine-breakthrough infections.
In unvaccinated individuals, variant of Concern (VOC) Alpha, Delta, and Omicron respiratory samples were found to have higher within-host diversity and were under neutral to purifying selection at the full genome level compared to non-VOC SARS-CoV-2. Breakthrough infections in 2-dose or 3-dose Comirnaty and CoronaVac vaccinated individuals did not increase levels of non-synonymous mutations and did not change the direction of selection pressure. Vaccine-induced antibody or T cell responses did not appear to have significant impact on within-host SARS-CoV-2 sequence diversification.
Our findings suggest that vaccination does not increase exploration of SARS-CoV-2 protein sequence space and may not facilitate emergence of viral variants.
Introduction
The SARS-CoV-2 pandemic continues to spread globally. Despite the vaccination of over 69% of the world population1, the risk of SARS-CoV-2 reinfections and breakthrough infections is increasing due to the emergence of new viral variants2,3. Multiple variants of concern (VOC) have demonstrated the ability to evade naturally-acquired or vaccine-induced immunity4,5,6. Therefore, it is crucial to investigate the impact of vaccination on the mutational and evolutionary processes of SARS-CoV-2.
Genomic surveillance has been used to trace the transmission and evolution of SARS-CoV-2 mutations at local, regional, and global scales throughout the pandemic7,8,9. However, there is still limited knowledge of how these mutations originate and accumulate within hosts. Within-host mutations can arise through replication errors or RNA damage/editing10 and they may be subject to fixation by stochastic (genetic drift) and deterministic (natural selection) processes. We and others have previously found that the SARS-CoV-2 transmission bottleneck between hosts is narrow8,11,12,13,14, suggesting that only few virions are transferred from the host during transmission. Most of the low-frequency mutations are not transmitted between patients, which constrains the use of intrahost single nucleotide variants (iSNVs) for effective contact tracing12,15,16. However, it remains important to investigate the within-host diversity of SARS-CoV-2 to understand host-level evolutionary forces.
Studying SARS-CoV-2 within-host diversity under different conditions may reveal factors that control virus evolution. Host and viral factors can both contribute to within-host diversity. Host factors such as species (animals/humans)17, viral shedding time18, and immune status19 were previously reported to have effects on intrahost SARS-CoV-2 diversity. It was hypothesized that prolonged infections in hosts with distinct immunological backgrounds (e.g., animals or immunocompromised patients) may hasten viral evolution and lead to the emergence of novel variants17,20. However, there is limited knowledge about post-vaccination characteristics of within-host selection pressures, which consistently act on the virus during the entire course of breakthrough infection. Besides, viral factors such as different virus lineages may also affect SARS-CoV-2 replication properties. SARS-CoV-2 VOCs have exhibited varying capacities to evade immunity4,6 and acquire higher transmissibility21,22. However, it is not clear whether different SARS-CoV-2 variants differ in within-host selection pressures.
Here, we analysed 2,820 deep-sequenced SARS-CoV-2 samples collected in Hong Kong (HK) between mid-2020 and 2022. The within-host diversity in SARS-CoV-2 infections from different lineages (VOCs and non-VOCs) and in breakthrough (Delta or Omicron) infections after Comirnaty or CoronaVac vaccination (two or three doses) were studied. Our results provide insights into the variation of within-host diversity, and the mutational patterns and selection pressures acting on viruses.
(SNIP)
Discussion
(Excerpt)
Vaccination is another factor which may affect the within-host virus evolution. We studied samples from Comirnaty and CoronaVac vaccine breakthrough infections and found that vaccination may be associated with changed mutation rates but might not change selection pressure. We found 2-dose Comirnaty vaccination was associated with increased synonymous nucleotide diversity and marginally significant purifying selection pressure at the full genome level, while similar effects of 2-dose CoronaVac vaccination was not as significant. Notably, the increased nucleotide diversity in specimens of Delta breakthrough infection in 2-dose Comirnaty vaccinated individuals is mostly synonymous rather than non-synonymous (Supplementary Table 4). Comirnaty vaccine is known to be more immunogenic than CoronaVac vaccine43 and this may contribute to our observation. It is also relevant to note that Comirnaty vaccine only has the spike protein as an immunogen but appears to impact on purifying selection elsewhere in the genome.
Crucially, vaccination does not increase exploration of the protein sequence space as non-synonymous nucleotide diversity does not seem to be increasing. For Omicron virus samples, we did not observe significant changes in incidence of iSNVs, nucleotide diversity or selection pressure in samples with 2-dose Comirnaty/CoronaVac vaccination. However, 3-dose Comirnaty vaccination virus samples seemed to have significantly lower incidence of iSNVs and nucleotide diversity than 2-dose or unvaccinated Omicron virus samples. The viral loads of 3-dose samples are similar to viral loads of other samples (Supplementary Fig. 13), suggesting that vaccination did not directly suppress viral replication, but might have limited exploration of sequence space. The low diversity observed in 3-dose Comirnaty Omicron samples was not as significant in 3-dose CoronaVac Omicron samples (there was no significant difference between collection lags or time since last dose between the two vaccines), possibly due to immunogenic difference between the two vaccines. We, however, do not exclude alternative hypotheses to explain this observation. Furt
As HK used an elimination strategy to control COVID-19, the individuals investigated in our study can be reliably categorised as immunologically naïve or vaccinated individuals, which is a significant advantage of our study. Nonetheless, our study has several limitations. Most of the studied cases have only single-timepoint samples, making it hard to study the temporal changes of within-host selection pressures. Although we made an effort to account for biases from sampling and different viral loads, false positive variant calls can still be an issue in analysing next-generation sequencing data, particularly from clinical samples that are of limited availability. A well-planned cohort, which can control major potential confounding factors, e.g., different demographic backgrounds, vaccination time lags, sample collection time points, and sequencing conditions, would provide a more robust estimation on iSNVs profiles in SARS-CoV-2 infections. Besides, in studying the effect of T cell pressure on within-host viral evolution, we could not perform an individual-based analysis since HLA typing of the patients was not possible in our study. In addition, since most of the individuals in our study were either infection naïve or vaccinated prior to infection, the effect of hybrid immunity on SARS-CoV-2 within-host evolution could not be addressed and requires further investigation.
In conclusion, our work suggests that SARS-CoV-2 within-host evolution may exhibit different patterns in different virus lineages and in vaccinated individuals. We found that 2-dose or 3-dose Comirnaty and CoronaVac COVID-19 vaccination does not seem to increase non-synonymous mutations in VOCs, suggesting that vaccination may limit the exploration of protein sequence space and the emergence of more viral variants.
(Continue . . . )
Gu, H., Quadeer, A.A., Krishnan, P. et al. Within-host genetic diversity of SARS-CoV-2 lineages in unvaccinated and vaccinated individuals. Nat Commun 14, 1793 (2023). https://doi.org/10.1038/s41467-023-37468-y